Compute Multi-trait GWAS with JASS
Once the GWAS summary statistics are integrated in the inittable (see Data preparation), you can generate multi-trait GWAS for any set of traits and for several joint tests with the command jass create-project-data (see command line reference for the detail of arguments).
Command Line example
Here is a mock up example of a command line to generate a multi-trait GWAS on 4 traits using the Omnibus test. See command line usage for more details
jass create-project-data --init-table-path init_table/init_table.hdf5 --phenotype z_MAGIC_GLUCOSE-TOLERANCE z_MAGIC_FAST-GLUCOSE z_MAGIC_FAST-INSULIN z_MAGIC_HBA1C --worktable-path ./work_glycemic.hdf5 --manhattan-plot-path ./manhattan_glycemic.png --quadrant-plot-path ./quadrant_glycemic.png
Generated Results
Whatever the test used, the command will generate three outputs:
A HDFStore containing several tables (Each table can be extracted using the can extracted to a tsv using the jass extract-tsv be read from the HDFStore with the pandas.read_hdf function):
'SumStatTab' : The results of the joint analysis by SNPs
'PhenoList' : the meta data of GWAS included in the multi-trait GWAS
'COV' : The H0 covariance used to perform joint analysis
'GENCOV' (If present in the initTable): The genetic covariance as computed by the LDscore.
'Regions' : Results of the joint analysis summarised by LD-independent regions (notably Lead SNPs by regions)
'summaryTable': a double entry table summarizing the number of significant regions by test (univariate vs joint test)
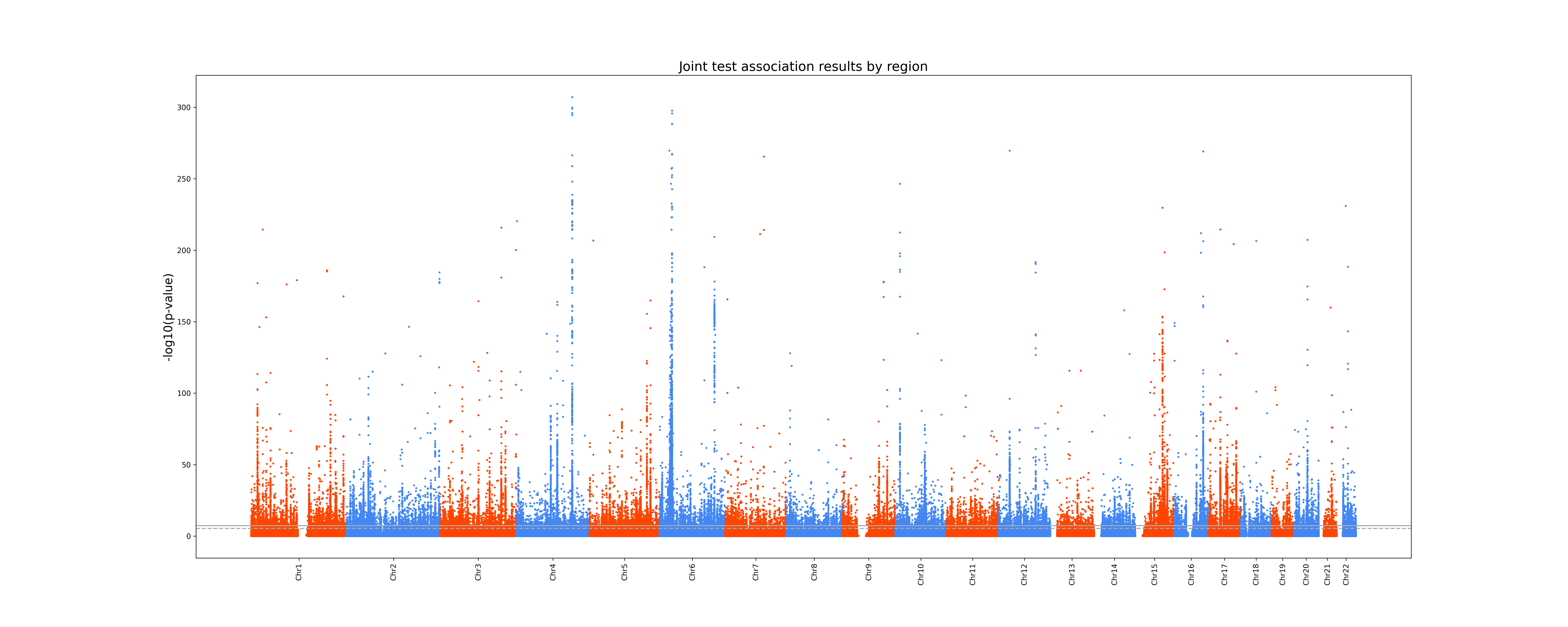
A .png Manhattan plot of the joint test p-values:
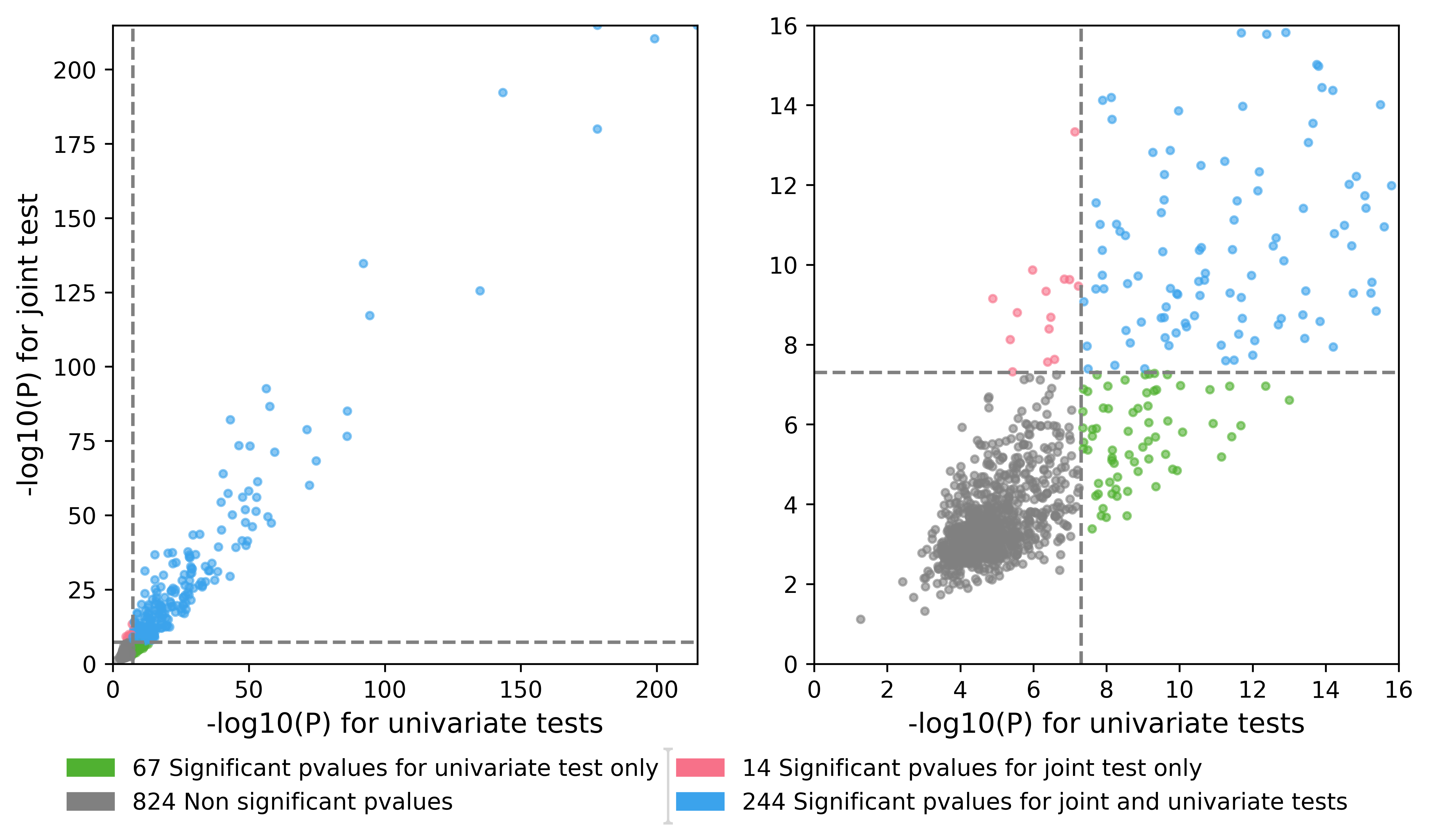
A .png Quadrant plot which is a scatter plot of the minimum p-value by region of the joint test with respect to the minimum p-value by region of the univariate tests. This plot provides an easy way to see if your joint analysis detected association not previously reported in the litterature.
Available multi-trait tests:
Several multi-trait tests are available through the jass create-project-data command. The Omnibus test can be interpreted as a multivariate analysis of variance using alternative allele counts as a grouping variable. The SumZ test perform a linear combination of traits Z-scores and test this combination for association. Please refer to [JLM+21] for the full description and derivation of those test
The Omnibus tests
If no option is provided to specify the test, a Omnibus test analysis will be performed. For instance:
jass create-project-data --init-table-path inittable_name.hdf5 --phenotypes z_CONSORTIUM1_TRAIT1 z_CONSORTIUM1_TRAIT2 z_CONSORTIUM2_TRAIT1 --worktable-path worktable_name.hdf5 --manhattan-plot-path manhattan_name.png --quadrant-plot-path /quadrant_name.png --qq-plot-path QQplots_name.png
The SumZ test
If the flag --sumz is passed to the jass create-project-data a SumZ test will be performed. By default, all the traits will have the same weight in the linear combination.
If the user wishes to, they can specify a vector of weight by using the --custom-loadings option.
jass create-project-data --init-table-path inittable_Update_COVID19.hdf5 --phenotypes z_INFECTION_INFLUENZA z_INFECTION_EAR-INFECTIONS --worktable-path worktable_test_SumZ.hdf5 --manhattan-plot-path manhattan_SumZ_test.png --quadrant-plot-path quadrant_SumZ_test.png --qq-plot-path QQplots_SumZ_test.png --sumz --custom-loadings test_loadings.csv"
the test_loadings.csv is comma separated file with the following structure:
trait |
weight |
|---|---|
z_INFECTION_INFLUENZA |
0.8 |
z_INFECTION_EAR-INFECTIONS |
-0.2 |
Access HDFStore components
Each table of the HDFStore is accessible through the command line tool jass extract-tsv (see command line reference for complete details).
jass extract-tsv --hdf5-table-path ./initTable.hdf5 --tsv-path './test_extract.tsv' --table-key SumStatTab
Alternately, you can use directly pandas read_hdf functions :
For instance if you want to access the Regions table :
pd.read_hdf("WK_test.hdf5", "Regions")
Note that is you wish that the SumStatTab table to be saved as a csv file you can provide the command lines with the --csv-file-path option and a csv will be generated as well. Outputting a csv will lengthen the execution and require the appropriate storage space (several 10Gb depending of the number of traits).